Biased cap-snatching of host RNA sequences

</br>
</br>
What is cap-snatching?
</br>
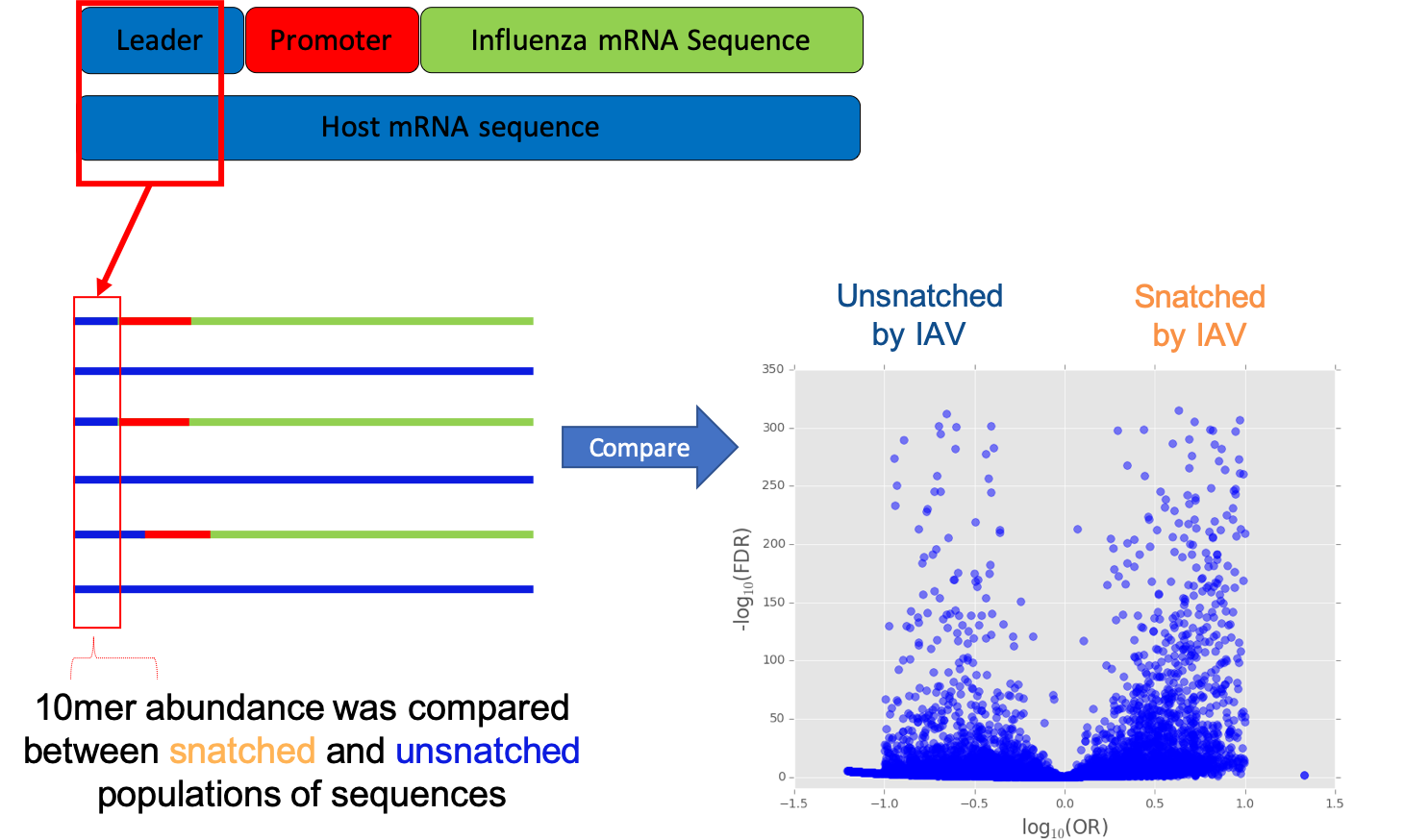
For this project we used CAGE sequencing to look at the 5’ ends of mRNA in primary human macrophages infected with influenza A H3N2 (IAV). We developed a method, to compare the ‘snatched’ population of sequences to the ‘unsnatched’ population. Our method allows us to compare the number of times a sequence is snatched or not snatched to the number of times it occurs in the cell.
</br>
</br>
Once we identified what sequences were being snatched, and those that were not being snatched we assigned gene names. This was not easy as our sequences were only 10 nucleotides long. We then used a method called Fast Gene Set Enrichment to look for pathways that were over-represented in our snatched and unsnatched sequences, taking into account the background sequences present in the cell.
</br>

</br>
##Compare LPS and IAV treatment
</br>

This analysis gave us a huge dataset filled with information about the transcripts that are expressed while macrophages fight IAV. We clustered these transcripts based on their expression profiles and watched as the transcriptional landscape changed over 24 hours of infection.

</br>
We also wanted to see how different pathogens had different effects so we used a datset we have previously published on to look at how bacterial (LPS) and viral (IAV) pathogens caused differential gene expression in our cells.

</br>
</br>
</br>
</br>
</br>
(minivirus by Claire Duggan)