Translational genomics in critical illness
We use computational biology and genomics to understand the mechanisms that make people desperately sick in intensive care, so that we can find ways to help them survive and recover. Our focus is sepsis, viral lung infections, and the impact of a shortage of oxygen. It’s quite old now, but the broad approach we take is summarised in this perspective article: Baillie JK, Science, 2014.
Work with us
We’ve been very lucky to make several contributions that have already improved medical practice. That is primarily because we’ve had some extremely capable and motivated people in the lab over the years. Many of them have moved on to greater things so, of course, we always need to find more. This website is aimed at people who might want to join us. If you’re excited about any of our work, even if we don’t have a post open at the moment, we want to hear from you.
You can get an idea of the kind of work we’ve done, and where it’s going, from the project summaries below.
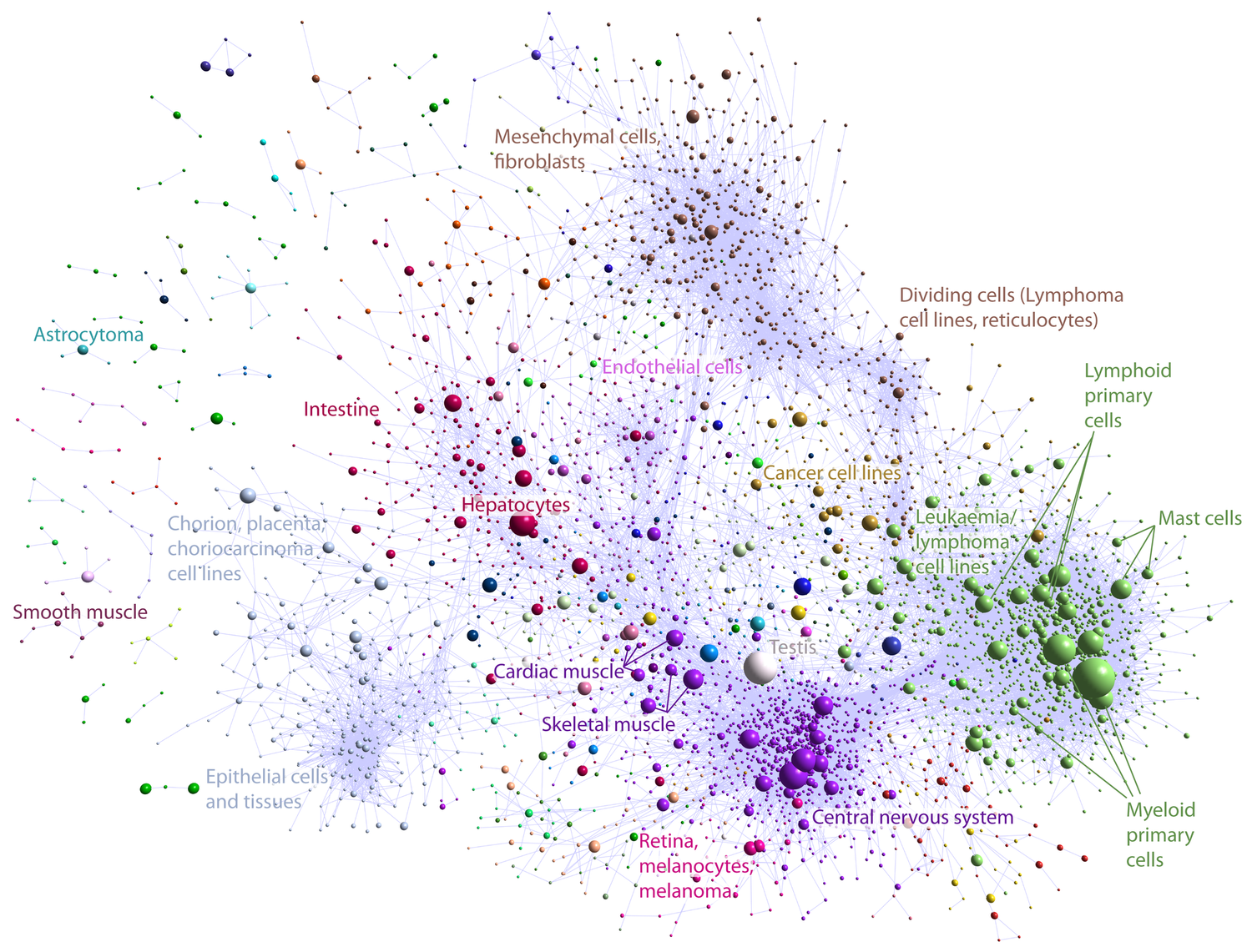
Genome-wide unbiased annotation of regulatory elements Nature, 2014
Systems biology
Computational biology is the main focus of the lab. Put simply, we’re trying to explain the molecular mechanisms underlying diseases in patients. We run a large-scale programme to discovery molecular consequences of genetic variation (molQTL). In order to test our predictions, we have a longstanding programme using genome editing in human and porcine cells and tissues to validate potential future drug targets.
Host Genomics
Host genetics provides the foundation for causal inference in our work. Since 2015 we have run the GenOMICC study to discover new human genetic associations with susceptibility to, and outcome from, critical illness. In 2020, only 5 months after the first Covid-19 patient recruited, we reported a functional genomic analysis in the GenOMICC that suggested a specific drug, baricitinib, would be an effective treatment for critical Covid-19. We went on to show that the treatment was effective in a clinical trial - to our knowledge, the first time this genetics-to-drug-treatment journey has been completed in any infectious disease or critical illness.

Interpretation of molecular mechanisms explaining genetic associations in Covid-19. Nature, 2023
Chord diagram showing weighted contributions of each data source about genes implicated in Covid-19 (Nature 2020)
MAIC (Meta-Analysis by Information Content)
We have developed a new computational approach to do something that is intuitive to biologists, but difficult for computers: to integrate data from diverse sources, weight it according to quality and relevance, and combine it. Our meta-analysis by infomation content (MAIC) algorithm does this, and opens up a range of opportunities across the field of genomics and biology.
You can learn more about the algorithm, and how to use it, at our MAIC project page
Hypoxia
Our theme of hypoxia research follows on from my early career work in high altitude medicine. In 2000 I set up a charity, Apex (altitude physiology expeditions), and organised a series of research expeditions. You can read about the past expeditions, and future ones, at the Apex website. Today we focus on physiological modelling and functional genomics.
Our mathematical models of gas exchange are used for teaching all over the world, and were the foundation for our development of the S/F94 clincial endpoint.

Helium-dilution measurement of lung volumes at Chacaltaya laboratory, Bolivia.
Large scale projects
We run several large-scale studies, which you can read about more at these sites:
- The global GenOMICC Study
- The UK Molecular Mechanisms Cluster
- ISARIC Comprehensive Clinical Characterisation Study
- The UK Outbreak Data Analysis Platform
Selected Publications (†joint authorship)
Citations: 82300 h-index: 105 Clarivate highly-cited: 2022-present ORCID:0000-0001-5258-793X
Baillie JK , Angus D, Burnham K, Calandra T, Calfee C, Gutteridge A, Hacohen N, Khatri P, Langley R, Ma’ayan A, Marshall J, ..., Randolph AG. Causal inference can lead us to modifiable mechanisms and informative archetypes in sepsis. Intensive Care Medicine (2024); 50: 2031-2042. PMC7616750
Pairo-Castineira E , Rawlik K, Bretherick AD, Qi T, Wu Y, Nassiri I, McConkey GA, Zechner M, Klaric L, Griffiths F, ..., Baillie JK. GWAS and meta-analysis identifies 49 genetic variants underlying critical COVID-19. Nature (2023); doi:10.1038/s41586-023-06034-3
Ho A , Orton R, Tayler R, Asamaphan P, Herder V, Davis C, Tong L, Smollett K, Manali M, Allan J, ..., Thomson EC†. Adeno-associated virus 2 infection in children with non-A-E hepatitis. Nature (2023); doi:10.1038/s41586-023-05948-2
Russell CD , Lone NI, Baillie JK. Comorbidities, multimorbidity and COVID-19. Nature Medicine (2023); 29: 334-343. doi:10.1038/s41591-022-02156-9
Wang B , Law A, Regan T, Parkinson N, Cole J, Russell CD, Dockrell DH, Gutmann MU, Baillie JK. Systematic comparison of ranking aggregation methods for gene lists in experimental results. Bioinformatics (2022); doi:10.1093/bioinformatics/btac621
The RECOVERY Collaborative Group . Baricitinib in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial and updated meta-analysis. Lancet (2022); 400: 359-368. PMC9333998
Swets MC , Russell CD, Harrison EM, Docherty AB, Lone N, Girvan M, Hardwick HE, Visser LG, Openshaw PJM, Groeneveld GH, ..., Baillie JK. SARS-CoV-2 co-infection with influenza viruses, respiratory syncytial virus, or adenoviruses. Lancet (2022); doi:10.1016/S0140-6736(22)00383-X
Kousathanas A , Pairo-Castineira E, Rawlik K, Stuckey A, Odhams CA, Walker S, Russell CD, Malinauskas T, Wu Y, Millar J, ..., Baillie JK. Whole genome sequencing reveals host factors underlying critical Covid-19. Nature (2022); doi:10.1038/s41586-022-04576-6
Ulferts R , Marcassa E, Timimi L, Lee LC, Daley A, Montaner B, Turner SD, Florey O, Baillie JK, Beale R. Subtractive CRISPR screen identifies the ATG16L1/vacuolar ATPase axis as required for non-canonical LC3 lipidation. Cell Reports (2021); 37: 109899. doi:10.1016/j.celrep.2021.109899
Thwaites RS , Sanchez Sevilla Uruchurtu A, Siggins MK, Liew F, Russell CD, Moore SC, Fairfield C, Carter E, Abrams S, Short C, ..., Baillie JK, Openshaw PJ. Inflammatory profiles across the spectrum of disease reveal a distinct role for GM-CSF in severe COVID-19. Science Immunology (2021); 6: PMC8128298
Horby P , Lim WS, Emberson JR, Mafham M, Bell JL, Linsell L, Staplin N, Brightling C, Ustianowski A, Elmahi E, ..., Baillie JK, Haynes R, Landray MJ. Dexamethasone in Hospitalized Patients with Covid-19. The New England Journal of Medicine (2021); 384: 693-704. PMC7383595
Pairo-Castineira E , Clohisey S, Klaric L, Bretherick AD, Rawlik K, Pasko D, Walker S, Parkinson N, Fourman MH, Russell CD, ..., Baillie JK. Genetic mechanisms of critical illness in COVID-19. Nature (2020); 591: 92-98. doi:10.1038/s41586-020-03065-y
Docherty AB , Harrison EM, Green CA, Hardwick HE, Pius R, Norman L, Holden KA, Read JM, Dondelinger F, Carson G, ..., Baillie JK, Semple MG. Features of 20133 UK patients in hospital with covid-19 using the ISARIC WHO Clinical Characterisation Protocol: prospective observational cohort study. BMJ (Clinical Research Ed.) (2020); 369: m1985. PMC7243036
Li B , Clohisey SM, Chia BS, Wang B, Cui A, Eisenhaure T, Schweitzer LD, Hoover P, Parkinson NJ, Nachshon A, ..., Baillie JK, Hacohen N. Genome-wide CRISPR screen identifies host dependency factors for influenza A virus infection. Nature Communications (2020); 11: 164. PMC6952391
Baillie JK , Arner E, Daub C, De Hoon M, Itoh M, Kawaji H, Lassmann T, Carninci P, Forrest ARR, Hayashizaki Y, Faulkner GJ, ..., Hume DA. Analysis of the human monocyte-derived macrophage transcriptome and response to lipopolysaccharide provides new insights into genetic aetiology of inflammatory bowel disease. PLoS Genetics (2017); 13: e1006641. PMC5358891
Baillie JK . Translational genomics. Targeting the host immune response to fight infection. Science (New York, N.Y.) (2014); 344: 807-8. doi:10.1126/science.1255074
Baillie JK† , Forrest ARR†, Kawaji H†, Rehli M†, de Hoon MJL, Haberle V, Lassmann T, Kulakovskiy IV, Lizio M, Itoh M, ..., Hayashizaki Y. A promoter-level mammalian expression atlas. Nature (2014); 507: 462-70. PMC4529748
Dunning JW , Merson L, Rohde GGU, Gao Z, Semple MG, Tran D, Gordon A, Olliaro PL, Khoo SH, Bruzzone R, ..., Baillie JK. Open source clinical science for emerging infections. The Lancet. Infectious Diseases (2014); 14: 8-9. PMC7158987
Everitt AR , Clare S, Pertel T, John SP, Wash RS, Smith SE, Chin CR, Feeley EM, Sims JS, Adams DJ, ..., Baillie JK, Walsh TS, Hume DA, Palotie A, Xue Y, Colonna V, Tyler-Smith C, Dunning J, Gordon SB, Smyth RL, Openshaw PJ, Dougan G, Brass AL, Kellam P. IFITM3 restricts the morbidity and mortality associated with influenza. Nature (2012); 484: 519-23. PMC3648786
Baillie JK† , Barnett MW†, Upton KR†, Gerhardt DJ, Richmond TA, De Sapio F, Brennan PM, Rizzu P, Smith S, Fell M, ..., Faulkner GJ. Somatic retrotransposition alters the genetic landscape of the human brain. Nature (2011); 479: 534-7. PMC3224101